
Steroid Hormone Signaling in Regulation of Oocyte Development
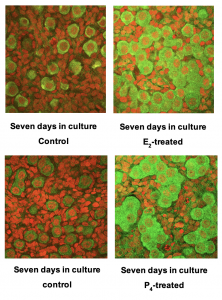
Our objective is to understand the role of steroid hormone signaling in cyst breakdown and oocyte survival. Although the mechanisms that control breakdown of cysts into individual oocytes and selection of some oocytes for survival are currently unknown, some evidence suggests a role for steroid hormones. Treatment of neonatal mice with natural estrogens such as estradiol (E2), or the phytoestrogen genistein or synthetic estrogens results in abnormal multiple oocyte follicles (MOFs). For genistein, this effect is mediated through Estrogen Receptor-b (ER-b). Recent data demonstrates that genistein treatment promotes oocyte survival and inhibits cyst breakdown leading to MOFs. However, the primary estrogen present in the maternal circulation is E2. Neonatal E2 treatment is known to cause MOFs in the adult ovary. Additionally, neonatal E2 treatment has been shown to inhibit cyst breakdown. Furthermore, progesterone (P4) treatment in organ culture inhibits cyst breakdown. Our model is that the removal of E2 and P4 from the fetal ovary results in triggering cyst breakdown and primordial follicle formation. Recent evidence has shown that blocking maternal estrogen synthesis promotes cyst breakdown, suggesting that steroid hormones are sourced from the maternal bloodstream.
Signaling Pathways in Regulation of Oocyte Development
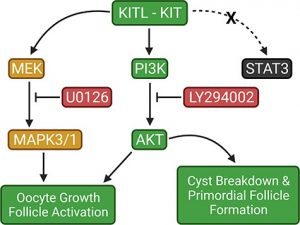 Our objective is to understand how the role of cell proliferation pathways may promote cyst breakdown and oocyte survival. Evidence suggests a role in specific cell pathways including the KIT tyrosine pathway. The KIT ligand is able to bind to KIT, which homodimerizes and autophosphorylates at tyrosine vesicles. This activates several downstream pathways including JAK and STAT, PI3K and MAPK pathways. Recent data has demonstrated an increase in follicle formation when neonatal mouse ovaries are cultured with KIT ligand and a decrease in follicle formation when ovaries are cultured with ACK2, which blocks the KIT receptor. Furthermore, the reduction of KITL expression results in primordial germ cell migration to ectopic sites and reduction in germ cell proliferation and survival. Together these data suggest that activation of the KIT pathway promotes primordial follicle formation. Current studies aim to promote follicle formation by culturing ovaries with different concentrations of the KIT ligand specifically to visualize the overall effects of KIT signaling on ooctye development.
Our objective is to understand how the role of cell proliferation pathways may promote cyst breakdown and oocyte survival. Evidence suggests a role in specific cell pathways including the KIT tyrosine pathway. The KIT ligand is able to bind to KIT, which homodimerizes and autophosphorylates at tyrosine vesicles. This activates several downstream pathways including JAK and STAT, PI3K and MAPK pathways. Recent data has demonstrated an increase in follicle formation when neonatal mouse ovaries are cultured with KIT ligand and a decrease in follicle formation when ovaries are cultured with ACK2, which blocks the KIT receptor. Furthermore, the reduction of KITL expression results in primordial germ cell migration to ectopic sites and reduction in germ cell proliferation and survival. Together these data suggest that activation of the KIT pathway promotes primordial follicle formation. Current studies aim to promote follicle formation by culturing ovaries with different concentrations of the KIT ligand specifically to visualize the overall effects of KIT signaling on ooctye development.
Additionally, some evidence suggests that insulin plays a role in follicle development, its role in follicle formation is not yet well understood. Like KIT, insulin is also capable of activating the PI3K pathway which is important for cell proliferation and differentiation. Therefore, our objective is to establish a role for insulin in primordial follicle formation. Current studies aim to create a mouse model of gestational diabetes by injecting pregnant females with streptozotocin (STZ). STZ causes damage to the insulin secreting cells, pancreatic beta cells, thereby creating a hypoinsulinemic and hyperglycemic environment in the dam. Glucose, but not insulin, is capable of crossing the placental barrier, thus we predict that the increased glucose levels in the embryos will cause an increase in insulin production. This model offers an opportunity to study the impact if increased glucose and insulin levels in the fetal ovary, as well as the effect of gestational diabetes on fetal oogenesis.